- Methodology
- Open access
- Published:
Protocol: genetic transformation of the fern Ceratopteris richardii through microparticle bombardment
Plant Methods volume 11, Article number: 37 (2015)
Abstract
Background
The inability to genetically transform any fern species has been a major technical barrier to unlocking fern biology. Initial attempts to overcome this limitation were based on transient transformation approaches or achieved very low efficiencies. A highly efficient method of stable transformation was recently reported using the fern Ceratopteris richardii, in which particle bombardment of callus tissue achieved transformation efficiencies of up to 72%. As such, this transformation method represents a highly desirable research tool for groups wishing to undertake fern genetic analysis.
Results
We detail an updated and optimized protocol for transformation of C. richardii by particle bombardment, including all necessary ancillary protocols for successful growth and propagation of this species in a laboratory environment. The C. richardii lifecycle comprises separate, free-living gametophyte and sporophyte stages. Callus is induced from the sporophyte apex through growth on cytokinin-containing tissue culture medium and can be maintained indefinitely by sub-culturing. Transgene DNA is introduced into callus cells through particle bombardment, and stable genomic integration events are selected by regeneration and growth of T0 sporophytes for a period of 8 weeks on medium containing antibiotics. Selection of T1 transgenic progeny is accomplished through screening T1 gametophytes for antibiotic resistance. In many cases sexual reproduction and development of transgenic embryos requires growth and fertilization of gametophytes in the absence of antibiotics, followed by a separate screen for antibiotic resistance in the resultant sporophyte generation.
Conclusions
Genetic transformation of C. richardii using this protocol was found to be robust under a broad range of bombardment and recovery conditions. The successful expansion of the selection toolkit to include a second antibiotic for resistance screening (G-418) and different resistance marker promoters increases the scope of transformations possible using this technique and offers the prospect of more complex analysis, for example the creation of lines carrying more than one transgene. The introduction of a robust and practicable transformation technique is a very important milestone in the field of fern biology, and its successful implementation in C. richardii paves the way for adoption of this species as the first fern genetic model.
Background
Modern plant developmental genetics is often underpinned by the creation and study of transgenic lines to test gene function in planta. Stable genetic transformation of ferns has proved challenging in the past, and as such our understanding of fern development lags significantly behind almost all other land plant lineages despite adoption of the fern species Ceratopteris richardii for laboratory use [1]. The fern family is extremely diverse, comprising at least 10,000 species, and has a global geographic range across which species frequently compete successfully with angiosperms [2]. Given that some fern species are now recognized as having significant commercial and industrial value (e.g. the arsenic-hyperaccumulating fern Pteris vittata is currently being exploited for remediation of arsenic-contaminated soils [3]), interest in understanding fern developmental genetics is increasing.
Ferns demonstrate an unusual lifecycle in that both the gametophyte (Figure 1a–d) and sporophyte stage (Figure 1e−g) grow as independent organisms, presenting a wide range of target stages for transformation. Whilst direct uptake of both RNA and DNA by germinating haploid fern spores [4–6], and particle bombardment of developing gametophytes [7] were successfully demonstrated as methods of transient transformation, until recently a viable method for the creation of stable transgenics proved elusive. P. vittata and a close relative of C. richardii, C. thalictroides, were successfully transformed by Agrobacterium tumefaciens infection of germinating spores [8], but transformation efficiencies were very low (0.053 and 0.03%, respectively). An alternative method utilizing particle bombardment of sporophyte-derived callus tissue (Figure 1h–j) [9] has since been developed, achieving more practical transformation efficiencies for both C. thalictroides and C. richardii (up to 86 and 72%, respectively). Since its first publication this method has undergone further testing and optimization to streamline the procedure for routine laboratory use with C. richardii. Here we present a detailed step-by-step protocol of this optimised method to allow adoption of this transformation technique by researchers not currently familiar with handling ferns.

The lifecycle of Ceratopteris richardii and its relation to microparticle-mediated transformation. The C. richardii lifecycle comprises two separate, free-living stages, the haploid gametophyte (a–d) and the diploid sporophyte (e–g). Dispersed as haploid spores (a), these germinate and develop rhizoids and a two-dimensional thallus (b), assuming either hermaphrodite (c) or male (d) characteristics by sexual maturity (approximately 9–11 days after germination). Fertilization of gametes results in the development of sporophyte embryos on the hermaphrodite thallus (e), which continue to develop independently of the gametophyte, producing first simple fronds (f) and then fronds of increasing size and complexity until the mature, spore-bearing stage is achieved (g). Genetic transformation of C. richardii is performed on the sporophyte (h–j). Young sporophytes are treated with cytokinin (CK) to induce callus tissue at the shoot apex (h) in place of new fronds (f) [9]. This callus is isolated in tissue culture and transformed through particle bombardment (i) of plasmid DNA carrying the transgene of interest and a linked antibiotic resistance marker. Stable transgene integrations into the C. richardii genome are identified by screening for antibiotic resistance during sporophyte regeneration (j): antibiotic is added to the media, killing untransformed callus cells. Regenerated transformant shoots (designated T0) can then be grown to maturity and transgenic T1 spores (the progeny) harvested.
Materials
Reagents
-
Ceratopteris richardii gametophytes and sporophytes are grown in tissue culture on C-fern nutrient [10] 1% agar medium, pH 6.0. This media can be bought commercially or prepared from stock solutions as described in [11] (Additional file 1). Sterile tissue culture plates of 90 mm diameter are used, requiring 25 ml of medium per plate.
-
Callus and regenerating sporophytes are grown in tissue culture on 1× Murashige and Skoog (MS) 2% sucrose 0.7% agar medium, pH 5.8 (MS-sucrose medium). Sterile tissue culture plates of 90 and 50 mm diameter are both used, requiring 25 and 15 ml of medium per plate, respectively.
-
Sporophytes are grown to maturity on soil (potting compost, e.g. Levingtons F3 soil mix).
-
Callus is generated and maintained by application of the cytokinin (CK) analogues 6-benzylaminopurine (BAP) or kinetin (KT) in MS-sucrose medium at 5 μM final concentration. Selection for stable transformants is carried out adding antibiotic (hygromycin B or the kanamycin analogue G-418) to MS medium at the required concentration (Table 1). Stocks of 5 mM BAP, 5 mM KT, 40 mg/ml hygromycin B and 50 mg/ml G-418 can be prepared in deionised sterile water, filtered through a 0.2 μm filter and stored indefinitely at −20°C.
Table 1 Antibiotic selection conditions for C. richardii transformation -
Callus is bombarded using tungsten microparticles (average diameter ≈ 1.3 μm) labelled with 5 μg of purified plasmid DNA at a concentration of 1 μg/μl. Microparticle labelling requires sterile 50% glycerol, 2.5 M CaCl2 and 0.1 M spermidine (each sterilised through a 0.2 μm filter), and 70 and 100% ethanol (EtOH).
-
Ceratopteris richardii is propagated by spores, which are available commercially and from various research laboratories. This protocol is optimised for use with the Hn-n strain, and has not been trialled on other strains.
-
Spores are sterilised prior to use with a mixture of sodium hypochlorite solution (2% chlorine) and Tween-20 (0.1% v/v).
-
Cellophane discs (90 mm diameter) (AA Packaging Ltd., UK).
-
Filter paper (90 mm diameter).
-
Sterile 90 mm diameter and 50 mm diameter tissue culture plates.
-
Sterile 2 ml microcentrifuge tubes.
-
Sterile Pasteur pipettes.
-
General reagents for tissue culture e.g. 70% EtOH, parafilm, micropore tape.
Equipment
-
Growth of C. richardii requires continuous high humidity and high temperature. It is recommended that controlled environment facilities are used that are capable of maintaining constant ≈ 85% humidity with a long day (LD) growth regime of 16 h light/8 h dark, 28°C/28°C. Controlled humidity is not strictly required for growth of gametophytes, callus or regenerating sporophytes in tissue culture.
-
Biolistic particle delivery system (e.g. Bio-Rad PDS-1000/He™) and associated reagents.
-
Class 1 laminar flow tissue culture cabinet.
-
Sterile forceps. Whilst watchmakers’ forceps are recommended for the transfer and manipulation of sporophytes during tissue culture, bayonet forceps are recommended to make the sterile transfer of callus tissue easier.
-
Platform vortexer.
-
Centrifuge.
Protocol
The protocol for transformation of C. richardii comprises the following steps: (1) generation of callus tissue from the sporophyte apex, (2) transgene introduction via particle bombardment, (3) selection of stable transformation events and regeneration of T0 sporophytes through antibiotic selection, and (4) establishment of transgenic lines from the T1 progeny. This last stage is necessary because in proof-of-principle experiments, T0 transgenic shoots were frequently found to be chimeric [9] and thus not all T1 progeny will carry the desired transgene. Whilst this protocol was found to achieve robust transformation efficiencies under a range of experimental conditions in our hands, the majority of steps detailed in this protocol are involved with tissue culture, and many are performed under sterile conditions. Therefore, prior practical experience of tissue culture techniques will make adoption of this protocol easier, and presumably improve the results obtained using this protocol in the short term. The time intervals given in this protocol reflect a practiced user in an environment where the system is already established.
Plasmid design: antibiotic resistance selection markers
In addition to the desired transgenic construct, plasmids for transformation of C. richardii require a linked antibiotic resistance selection marker under a constitutive promoter. C. richardii callus is susceptible to hygromycin B [9] and the kanamycin analogue G-418 (Additional file 2); the genes hygromycin B phosophotransferase (HygR) [9] and Neomycin phosphotransferase II (NPTII) (Additional file 3) can both be used as selection markers. The commonly-used viral 35S promoter drives sufficient expression in C. richardii for use with a selectable marker [9]. Similarly, the nopaline synthase (nos) promoter (295 bp) in the Gateway binary vector system [12] is sufficient to confer resistance in test transformations (Additional file 3). However, a truncated nos promoter variant (184 bp) from the pART27 vector [13] failed to regenerate T0 shoots under antibiotic selection (Additional file 3). As such, not all existing plant vectors can be expected to function in C. richardii. Where possible, it is recommended that expression tags be included with the target transgene to allow rapid determination of functional transgene expression in T0 shoots and T1 transgenic lines: plasmid shearing during particle bombardment means that the presence of a resistance marker is not a guaranteed indicator of a linked transgene [14].
A. Callogenesis and callus maintenance
Callus tissue is generated from young sporophytes. Unlike seed plants, ferns propagate through haploid spores (Figure 1a), thus requiring germination and growth of the gametophyte phase in sterile tissue culture to obtain sporophytes for callogenesis. Initial stocks of sterile spores can be obtained commercially, or previously harvested spores can be sterilised prior to germination. Spores are stored dry at room temperature. A 20 mg aliquot of dry spores typically provides sufficient material for up to eight plates, although sufficient stocks of callus for transformation can ultimately be obtained from a single plate of spores.
-
1.
Culture gametophytes from spores under sterile conditions on 90 mm diameter C-fern medium tissue culture plates (see detailed protocols in Additional files 1, 4) until sexually mature (Figure 1c, d; typically 9–11 days after sowing).
-
2.
Add 4–5 ml sterile water to each plate to fertilize the gametophyte population and induce sporophyte development. Swirl the plate gently to distribute the water evenly, then reseal and return to incubator. To maximise the frequency of fertilization, repeat this step after 3–4 days. Subsequently, maintain high humidity within each plate by adding 2 ml sterile water every 4–5 days.
Note If fertilization is successful, sporophytes typically become visible approximately seven days after fertilization, growing on the gametophyte thallus and distinguishable as a darker green colour (Figure 1e).
-
3.
Sporophytes are suitable for callus induction once they have developed a visible shoot and root, typically between 10 and 14 days after fertilization (Figure 2a). Induce callus by transferring whole individual sporophytes to 1 × MS 2% sucrose 0.7% agar plates (pH 5.8) containing 5 μM BAP (MS-BAP medium). Seal the plates with micropore tape and incubate under 28°C LD growth conditions. After 10–14 days, callus tissue should become visible at the shoot apex in place of new fronds (Figure 2b).
Note In our hands C. richardii callus tissue developed poorly on C-fern medium [9], and as such it should not be used for callus induction or maintenance. The inclusion of auxin in the growth medium [9] was found to be unnecessary for both induction and maintenance of callus tissue (Additional file 5). Healthy callus suitable for transformation is typically emerald green in colour and friable.
Figure 2 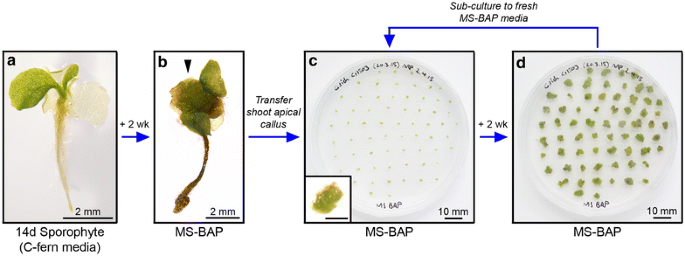
Callogenesis and callus maintenance. Callus tissue is induced from young sporophytes (a). Growing sporophytes on 1× MS 2% sucrose 0.7% agar medium (pH 5.8) containing 5 μM BAP (MS-BAP) causes development of callus at the shoot apex within 2 weeks (arrowhead b). Callus tissue is then removed from the progenitor sporophyte using forceps and transferred to fresh MS-BAP medium (c). Inset shows individual callus piece, scale 1 mm. Callus tissue on MS-BAP maintains an undifferentiated fate and will grow independently. After 2 weeks (d) callus is typically large enough for bombardment and/or sub-culturing to fresh medium to maintain callus stocks for future bombardments.
-
4.
Using sterile watchmakers’ forceps, excise small pieces of callus tissue from the apex and transfer to fresh MS-BAP plates (Figure 2c). Incubate under under 28°C LD growth conditions.
Note Callus plates do not require the addition of water to maintain callus growth.
-
5.
To maintain stable callus stocks in tissue culture, transfer small pieces from existing plates to fresh MS-BAP medium (sub-culturing) every 2 weeks (Figure 2d). At 2 weeks a single plate of callus is typically sufficient to prepare two bombardment plates (see step B1).
Troubleshooting If callus colonies become too large then tissue differentiation can occur. If the initial callus population shows strong signs of differentiation after separation from the sporophyte, sub-culture the callus after 1 week to maintain high penetration of BAP into the tissue.
Note Callus stocks can be maintained in tissue culture for over a year and remain competent for transformation [9], but in our experience the quality of regenerated T0 shoots that are recovered from transformation degrades over time with ageing callus stocks. To maintain maximum quality it is recommended that a fresh callus stock be generated every 3–4 months.

B. Callus transformation and regeneration of T0 transgenic sporophytes
Ceratopteris richardii callus tissue is transformed by particle bombardment. Proof-of-principle experiments found that some regenerated transgenic T0 shoots either do not carry or do not express the desired transgene [9], and these cannot be distinguished from fully-transgenic shoots when transplanting to soil. The bombardment and regeneration conditions outlined below have been optimised through tests using a 35S::GUS transformation marker to increase the proportion of genuinely transgenic sporophytes within the regenerated population (see Additional file 6). All of the following steps should be carried out under sterile conditions in a laminar flow cabinet, unless stated otherwise.
-
1.
Transfer callus from MS-BAP stock plates to 1xMS 2% sucrose 0.7% agar (pH 5.8) 5 μM KT (MS-KT) 50 mm bombardment plates (see Additional file 1). Callus pieces should be arranged very densely to create a compact circular target for bombardment approximately 30 mm in diameter (Figure 3a).
Note Bombardment plates can be prepared up to 2 days prior to bombardment, with further callus growth during this time increasing target density, but in this scenario plates should be sealed with parafilm and incubated at 28°C under LD conditions prior to bombardment.
Note Callus should not be bombarded or regenerated on media containing BAP, which blocks subsequent shoot regeneration even 6 weeks after BAP treatment has been removed (Additional file 6). Although not essential for successful regeneration, including KT in the media during bombardment and for 2 weeks afterwards marginally increases regeneration efficiency and the final size of regenerated shoots (Additional file 6).
Figure 3 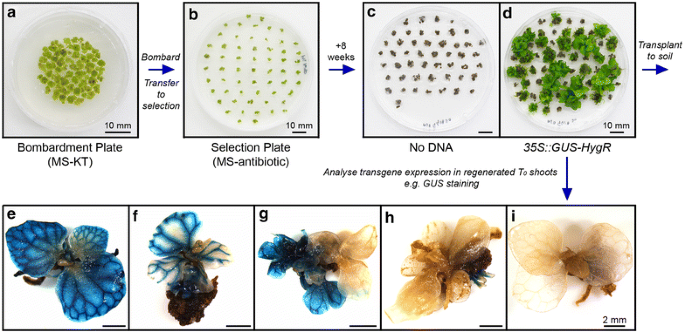
Callus bombardment and T0 sporophyte regeneration. Genetic transformation of C. richardii callus is through particle bombardment (a–d). Example shown is transformation of 35S::GUS-HygR. Callus is assembled for bombardment on a 50 mm plate containing 1× MS 2% sucrose 0.7% agar media (pH 5.8) containing 5 μM KT (MS-KT), creating a dense 25–30 mm diameter target for bombardment. After bombardment callus is transferred onto MS-KT plates containing antibiotic (b) and selection is maintained for 8 weeks, transferring to fresh selective media every 2 weeks. KT treatment is removed after 2 weeks to allow tissue differentiation. Untransformed callus is killed and turns dark brown without further growth (c). Transformed callus cells survive and divide, regenerating into differentiated sporophytes (d) suitable for transplanting to soil. Multiple sporophytes can regenerate from the same piece of callus. Transgene expression within regenerated sporophytes is variable (e–f), with some demonstrating full transformation (e), others apparently chimeric expression (f) and some demonstrating no expression despite antibiotic resistance (g–i).
-
2.
Prepare sterile tungsten microparticles and label with plasmid DNA for transformation [15] (see detailed protocol in Additional file 7). Sterilisation and labelling of microparticles can be conducted on the bench, outside of a laminar flow cabinet.
-
3.
Bombard callus tissue with the DNA of interest under sterile conditions, following the set-up and operating protocols specified by the manufacturer of the bombardment system.
In our hands Whilst successful transformation of callus occurred across the entire range of firing pressures tested [400–1,100 pounds per square inch (psi)] using the Bio-Rad PDS-1000/He™ system, bombardment at 600 psi resulted in the greatest degree of regeneration and confirmed transformation of T0 sporophytes (Additional file 6).
Note It is strongly recommended that at least one ‘No DNA’ control (unlabelled microparticles) be bombarded at the start of each transformation session to screen for successful antibiotic selection and for the presence of cross-contamination from prior bombardments. It is recommended that multiple bombardments of the same plasmid be performed to ensure the creation of independent transgenic lines. If multiple plasmids are being bombarded within the same session, it is further recommended that plasmids carrying different antibiotic selection markers be bombarded to minimize the chances of cross-contamination, and that a further No DNA control be bombarded between bombardments of the different plasmids.
-
4.
Re-seal bombardment plates with parafilm and incubate them under 28°C LD growth conditions for 1–3 days prior to transfer to selection media.
Note Whilst acceptable frequencies of regeneration and transformation were achieved after only an overnight recovery post-bombardment before moving callus onto selection, the greatest proportion of fully-transgenic sporophytes suitable for transplant to soil were obtained after a 3 days recovery interval (Additional file 6).
-
5.
Transfer callus onto MS-KT 90 mm selection plates containing antibiotic at the appropriate concentration (Table 1) and incubate plates under 28°C LD growth conditions. Individual callus pieces should be spaced separately (Figure 3b) to allow for ease of selection of independent regenerated shoots. Typically all callus tissue from one bombardment plate can fit onto a single 90 mm selection plate.
Important Selection plates MUST be sealed with parafilm to retain high humidity, otherwise shoot regeneration will fail.
-
6.
Maintain callus under continuous antibiotic selection for 8 weeks, transferring callus to fresh MS-antibiotic medium every 2 weeks. Regenerated sporophytes are typically large enough to transplant to soil by 8 weeks (20–30 mm rosette diameter, Figure 3d).
Note After the first 2 weeks KT should not be included in the regeneration medium: continued application of KT is detrimental to the quality of regenerated shoots (Additional file 6). Callus tissue typically begins turning brown by 2 weeks after the start of selection, and is entirely brown by 4–6 weeks (Figure 3c). Sporophyte regeneration is typically visible from 4 weeks as small green nodes growing from otherwise brown/black callus.
Note The continued growth of regenerating sporophytes can result in crowding. It is recommended that at 6 weeks after bombardment regenerating callus tissue from each bombardment be spaced over two selection plates instead of one. Frequently multiple sporophytes will regenerate from the same piece of callus. It is important for subsequent transgenic selection (step B7) that sporophyte clusters arising from the same callus not be broken up during selection.
Troubleshooting If selection has been successful, the accompanying No DNA control plates should show no evidence of sporophyte regeneration (Figure 3c). If callus tissue remains substantially green after 4–6 weeks selection, including the No DNA control, antibiotic selection is failing. Transfer callus to media prepared with a fresh antibiotic stock. If sporophyte regeneration occurs on the No DNA control plate in addition to the test bombardments, contamination with DNA from this or previous bombardments has probably occurred. Clean all biolistic gun components thoroughly prior to any repeat bombardments—it is recommended that No DNA bombardments be tested first to confirm the absence of contamination. If sporophyte regeneration has failed on all plates, check that the resistance marker for the plasmid in question is functional. If sporophyte regeneration persistently fails, check that regeneration conditions are correct and ensure that plates are being sealed with parafilm.
-
7.
Unseal tissue culture plates and transplant individual T0 sporophytes to soil using forceps (see detailed protocol in Additional file 4). Any T0 sporophytes not transplanted to soil can be analysed to assess transgene expression.
Important Multiple independent transgenic lines should be established for each transgene to obtain a reliable and accurate record of transgene expression and function. Sporophytes obtained from separate bombardments represent independent transformation events, but individuals from the same bombardment could be clones of the same initial genomic integration [9]. This probability increases in sporophytes regenerating from the same piece of callus. Proof-of-principle experiments found that a single regenerated population of T0 sporophytes contains a range of transgene expression patterns [9] (Figure 3e–i). Not all individuals regenerating from the same callus express the desired transgene when tested (Figure 3g–i), and transgene expression was not found to equate with regenerated sporophyte size. To maximise the number of independent lines obtained when transplanting sporophytes to soil, it is therefore recommended that only a single individual be transplanted from any particular callus cluster. In our experience, a relatively reliable indicator of genuine antibiotic resistance in a regenerated shoot is the growth of roots into the selection media.
-
8.
Grow T0 sporophytes to maturity on soil. Regenerated sporophytes take 2–3 months to begin producing reproductive fronds (Figure 1g) containing T1 spores.
-
9.
Harvest mature reproductive fronds from individual T0 sporophytes and collect T1 spores (see detailed protocol in Additional file 4). Take care to avoid cross-contamination between individuals when harvesting.
Note In our experience C. richardii spores retain dormancy for at least 1 month after harvesting, with very poor spore germination observed if fresh spores are imbibed during this time. An after-ripening period of at least 2 months after harvesting is therefore recommended to make screening of T1 lines more reliable.

C. Selection of T1 lines
Because of the apparently chimeric nature of many regenerated T0 sporophytes, not all T1 spores harvested carry the desired transgene. As such, each T1 line must be screened to identify transgenic individuals, the frequency of which can vary by a wide degree [9]. Furthermore, in proof-of-principle experiments antibiotic selection rendered even resistant T1 gametophytes sterile [9], necessitating the induction of T1 sporophytes in the absence of selection at the gametophyte stage, with subsequent identification of transgenic sporophytes through separate antibiotic resistance screening. Although some examples of transgenic sporophytes forming directly from transgenic gametophytes have since been observed (Additional file 8), this is not universal and potentially dependent on the selection marker used. The protocol described below assumes separate induction and screening of sporophyte populations for antibiotic resistance.
T 1 gametophyte screening
-
1.
Sterilise an aliquot of spores for each T1 line to be screened (see detailed protocol in Additional file 4) including an aliquot of untransformed wild type (WT) spores as a control. Incubate at room temperature in darkness for 3–5 days.
-
2.
Sow each spore line over two C-fern medium 90 mm diameter tissue culture plates, one without antibiotic selection and one containing antibiotic at the appropriate concentration (Table 1). Culture gametophytes under sterile conditions until 9–11 days old (see detailed protocol in Additional file 4).
Note If further analysis of resistant T1 gametophytes is required, e.g. GUS staining or fluorescence microscopy, their removal from the plate can be facilitated by placing a sterile cellophane disc over the medium prior to spore sowing (see detailed protocol in Additional file 4). It is recommended that cellophane discs not be used for control plates if sporophytes are to be induced and grown from them, as they interfere with root penetration into the growth medium.
-
3.
Screen T1 lines visually for antibiotic resistance. Control plates should carry developing gametophytes for all lines (Figure 4a, b). Antibiotic selection should kill all germinating untransformed spores [9], resulting in no visible gametophyte growth on the WT antibiotic selection plate (Figure 4c). Transgenic T1 gametophytes with antibiotic resistance should develop and be visible in the presence of antibiotic (Figure 4d).
Note After screening, resistant gametophytes can then either be analysed further for transgene expression (Figure 4e) or attempts can be made to induce sporophytes by adding sterile water to the plate.
Note Growth of gametophytes under antibiotic selection can be slower than under control conditions, and their precise developmental stage will need to be confirmed visually prior to screening.
Troubleshooting If gametophytes are developing on the WT antibiotic plate, selection has not worked effectively. The antibiotic stock may have become degraded, or contamination of the WT spore stock with transgenic spores might have occurred. Repeating the screen using a fresh antibiotic stock should determine this. If no gametophytes develop for a T1 line on either control or selective media, spore sterilisation might have been too severe or the spore stock may not be viable. Repeat the selection screen, sterilising spore stocks for a shorter period (5–10 min).
Figure 4 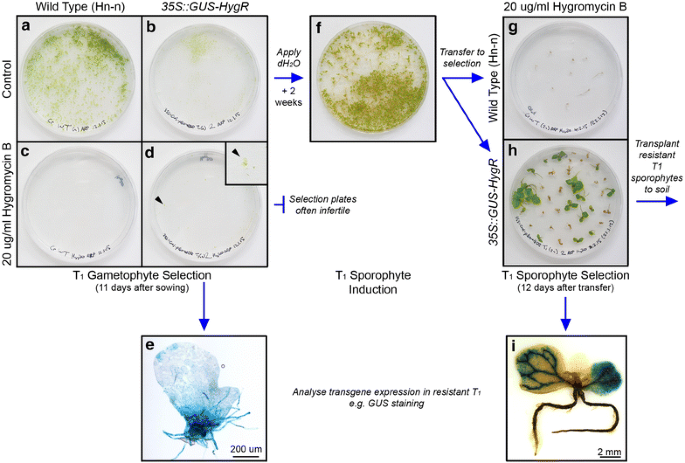
Selection of T1 transgenic lines. Transgenic T1 individuals are selected for inherited antibiotic resistance separately at the gametophyte (a–e) and sporophyte (f–i) stages. Example shown is 35S::GUS-HygR. a–e Gametophyte selection. Spores are sown on two C-fern 1% agar (pH 6.0) plates: one containing antibiotic (selective plate), the other not (control plate). By 11 days after sowing, WT (a) and T1 (b) gametophyte populations have developed on the control plates. Under antibiotic selection, WT gametophytes have not developed (c) but some resistant T1 gametophytes have developed successfully (d). Inset shows resistant gametophyte developing on the plate (arrowhead). Resistant T1 gametophytes can then be analysed for transgene expression in addition to antibiotic resistance (e). f–i Sporophyte selection. T1 sporophytes are induced from the control gametophyte plate by repeated application of sterile water. Sporophytes typically become visible on the plate within 2 weeks (f). Individual sporophytes are then transferred to C-fern 1% agar (pH 6.0) plates containing antibiotic to screen for antibiotic resistance. Selection typically requires 7–14 days. Antibiotic selection kills WT (g) and non-transgenic T1 sporophytes (h), whilst transgenic T1 sporophytes within the population survive and continue to grow (h). After antibiotic selection resistant T1 sporophytes can be transplanted to soil, or analysed for transgene expression in addition to antibiotic resistance (i).

T 1 sporophyte screening
-
4.
Induce sporophytes from the control plate of each line by adding sterile water (see step A2).
Note T1 sporophytes induced without selection from a mixed population of transgenic and non-transgenic gametophytes may or may not be transgenic, and so must also be screened for antibiotic resistance.
-
5.
When sporophytes are visible on the fertilized gametophyte plates (Figure 4f) transfer individual sporophytes to 90 mm C-fern plates containing antibiotic at the appropriate concentration (Table 1). After transferring, add 2 ml sterile water drop-wise to the plate with a sterile Pasteur pipette to increase humidity. Seal each plate with micropore tape and incubate under 28°C LD growth conditions. If plates become dry during incubation, repeat the addition of 2 ml sterile water every 4–5 days.
Note Approximately 25 sporophytes can be grown on a single selective plate. It is recommended that WT sporophytes be included in each selective screen for comparison against T1 sporophytes, and that some WT sporophytes be grown on media without antibiotic selection to provide comparisons against resistant T1 lines at future growth stages.
-
6.
Screen sporophytes visually for antibiotic resistance. Sporophytes lacking antibiotic resistance typically die 7–14 days after the start of selection (Figure 4g), whilst resistant sporophytes continue to grow (Figure 4h).
-
7.
Transplant resistant sporophytes to soil for further growth (see Additional file 4).
Note It is recommended that at least 4 resistant individual sporophytes from each T1 line be grown on soil to provide an assessment of the variability of transgene expression and genomic T-DNA insertions within that particular line. Any resistant T1 sporophytes not transplanted to soil can be analysed for transgene expression at this stage (Figure 4i).
Comments
As a transformation technique, particle bombardment has a number of disadvantages compared to techniques such as Agrobacterium-mediated transformation. Most notably, transgenics regenerated from particle bombardment typically contain a greater number of copies of the inserted transgene than those transformed by Agrobacterium [16]. Furthermore, physical shearing of T-DNA on impact with the plant cells results in the incorporation of fragmentary T-DNA, and the separation of the transgene cassette of interest from the antibiotic selection marker. All of these make the interpretation of any transgenic phenotypes and expression patterns more complex. As a consequence, it is important that any C. richardii transgenic lines produced through particle bombardment be evaluated thoroughly for copy number and the presence of full-length T-DNA. This can be achieved in the T1 generation through genotyping PCR and Southern analysis [8, 9]. If lines carrying single copy insertions are not identified in the T1 generation, T-DNA copy number can be reduced in subsequent generations by back-crossing to WT: one of the advantages of C. richardii as an experimental system is that sexual reproduction occurs under tissue culture conditions and can thus be tightly controlled.
Particle bombardment is applicable to a broader range of plant species than Agrobacterium-mediated transformation, as many are not naturally susceptible to infection [17]. In the case of C. richardii, Agrobacterium infection of gametophytes resulted in transformation at low efficiencies with practical difficulties associated with selection of transgenics [8], and attempts to transform sporophytic callus tissue using Agrobacterium were unsuccessful [9].
Conclusions
Genetic transformation of C. richardii through particle bombardment was found to be robust under a broad range of bombardment and recovery conditions, and requires relatively little specialist knowledge to implement beyond familiarity with tissue culture. As such, this protocol represents a practicable method of generating transgenic lines in sufficient quantity for reliable evaluation of transgene function and establishing it in new labs should be relatively simple. The successful expansion of the selection toolkit to include a second antibiotic for resistance screening (G-418) increases the scope of transformations possible using this technique and offers the prospect of more complex genetic analysis, for example the creation of lines carrying more than one transgene. The introduction of a robust and practicable transformation technique represents a very important milestone in the field of fern biology, and its successful implementation in C. richardii paves the way for adoption of this species as the first fern genetic model.
Abbreviations
- BAP:
-
6-benzylaminopurine
- bp:
-
base pairs
- CK:
-
cytokinin
- EDTA:
-
ethylenediaminetetraacetic acid
- EtOH:
-
ethanol
- GUS:
-
β-glucuronidase
- He2 :
-
helium (gas)
- HygR:
-
hygromycin B phosphotransferase
- KT:
-
kinetin
- LD:
-
long day
- MS:
-
Murashige and Skoog
- nos:
-
nopaline synthase
- NPT:
-
neomycin phosphotransferase
- psi:
-
pounds per square inch
- rpm:
-
revolutions per minute
- S.E.:
-
standard error
- T-DNA:
-
transgene DNA
- WT:
-
wild type
References
Hickok LG, Warne TR, Fribourg RS (1995) The biology of the fern Ceratopteris and its use as a model system. Int J Plant Sci 156:332–345
Mehltreter K, Walker LR, Sharpe JM (2010) Fern Ecology. Cambridge University Press, New York
Kertulis-Tartar GM, Ma LQ, Tu C, Chirenje T (2006) Phytoremediation of an arsenic-contaminated site using Pteris vittata L.: a two-year study. Int J Phytoremediation 8:311–322
Hickok LG, Warne TR, Slocum MK (1987) Ceratopteris richardii: applications for experimental plant biology. Am J Bot 74:1304–1316
Klink VP, Wolniak SM (2001) Centrin is necessary for the formation of the motile apparatus in spermatids of Marsilea. Mol Biol Cell 12:761–776
Stout SC, Clark GB, Archer-Evans S, Roux SJ (2003) Rapid and efficient suppression of gene expression in a single-cell model system, Ceratopteris richardii. Plant Physiol 131:1165–1168
Rutherford G, Tanurdzic M, Hasebe M, Banks JA (2004) A systemic gene silencing method suitable for high throughput reverse genetic analysis of gene function in fern gametophytes. BMC Plant Biol 4:6
Muthukumar B, Joyce BL, Elless MP, Stewart N (2013) Stable transformation of ferns using spores as targets: Pteris vittata (Chinese brake fern) and Ceratopteris thalictroides (C-fern ‘Express’). Plant Physiol 163:648–658
Plackett ARG, Huang L, Sanders H, Langdale JA (2014) High-efficiency stable transformation of the model fern species Ceratopteris richardii via microparticle bombardment. Plant Physiol 165:3–14
Klekowski EJ (1969) Reproductive biology of the Pteridophyta. IV. A study of the Blechnaceae. Bot J Linn Soc 62:361–377
Hickok LG, Warne TR (1998) C-Fern manual. Carolina Biological Supply Company, Burlington, VT
Karimi M, Inzé D, Depicker A (2002) GATEWAY™ vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7:193–195
Gleave AP (1992) A versatile binary vector system with a T-DNA organisational structure conducive to efficient integration of cloned DNA into the plant genome. Plant Mol Biol 20:1203–1207
Hansen G, Wright MS (1999) Recent advances in the transformation of plants. Trends Plant Sci 4:226–231
Sanford JC, Smith FD, Russel JA (1993) Optimizing the biolistic process for different biological applications. Methods Enzymol 217:483–509
Travella S, Ross SM, Harden J, Evrett C, Snape JW, Harwood WA (2005) A comparison of transgenic barley lines produced by particle bombardment and Agrobacterium-mediated techniques. Plant Cell Rep 23:780–789
Altpeter F, Baisakh N, Beachy R, Bock R, Capell T, Christou P et al (2005) Particle bombardment and the genetic enhancement of crops: myths and realities. Mol Breed 15:305–327
Authors’ contributions
ARGP and JAL designed the protocol optimization experiments and drafted the manuscript. ARGP and EHR conducted the experimental work. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to Julie Bull and Mary Saxton for help with plant maintenance and technical support. This work was supported by an European Research Council Advanced Investigator Grant “Evolution and Development in Plants”.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Media for C. richardii tissue culture. Protocols for the preparation of C-fern and MS agar media used during C. richardii transformation.
Additional file 2:
G-418 selection conditions for C. richardii. Kill curves demonstrating the necessary concentration of G-418 antibiotic needed for selection of C. richardii callus, gametophytes and sporophytes.
Additional file 3:
Evaluation of antibiotic selection markers. Design of constructs used to test promoter function driving HygR and NPTII resistance markers, and the mean regeneration frequencies achieved.
Additional file 4:
Culturing protocols for C. richardii. Protocols for the preparation and growth of C. richardii in sterile tissue culture and on soil.
Additional file 5:
Evaluation of callogenic hormone treatments. Comparison of two hormone treatments (BAP + IBA vs. BAP only) on callus maintenance and callus transformation efficiency.
Additional file 6:
Optimisation of bombardment conditions. Results of test 35S::GUS-HygR transformation efficiencies under different bombardment conditions: KT treatment during regeneration, He2 firing pressure and recovery interval length.
Additional file 7:
Preparation of microparticles for bombardment. Protocols for the sterilisation and DNA-labelling of microparticles for bombardment.
Additional file 8:
Direct induction of transgenic sporophytes from gametophytes under antibiotic selection. An example of the successful generation of T1 sporophytes from gametophytes under antibiotic selection and subsequent confirmation of their transgenic status.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Plackett, A.R.G., Rabbinowitsch, E.H. & Langdale, J.A. Protocol: genetic transformation of the fern Ceratopteris richardii through microparticle bombardment. Plant Methods 11, 37 (2015). https://doi.org/10.1186/s13007-015-0080-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13007-015-0080-8